保持药物化学性质和功能一致性的基于自动编码器的药物-靶点相互作用预测
摘要
动机:探索潜在的药物-靶相互作用(DTI)是药物发现和再利用的关键步骤。近年来,通过计算方法预测可能的DTI逐渐成为研究热点。然而,以往的大多数研究都没有明智地考虑到药物的化学性质与其功能之间的一致性。这些关系的改变可能会对DTI的预测产生严重的负面影响。
结果:提出了一种基于自动编码器的空间一致性约束下的DTI预测方法AEFS。建立一个异构网络,集成药物、蛋白质和疾病信息。原始药物特征由多层编码器投影到嵌入(蛋白质)空间,再由解码器投影到标签(疾病)空间。在这个过程中,引入了药物的临床信息来辅助DTI的预测。AEFS通过保持药物相关性在原始特征空间、嵌入空间和标签空间中的分布,保持了药物化学性质和功能的一致性。实验比较表明,AEFS对非平衡数据具有更强的鲁棒性,在DTI预测中具有明显的优势。案例研究进一步证实了其挖掘潜在药物-靶相互作用的能力。
可用性:AEFS的代码可在https://github.com/JackieSun818/AEFS上获得
1 Introduction
药物可以通过药物-靶相互作用(DTI)增强或抑制相关目标蛋白的表达,从而影响疾病的状况(Olayan等人,2018年)。识别DTI是药物发现和药物再利用的关键步骤(Chen等人,2016;Chu等人,2020;Sun等人,2020;Li等人,2020)。然而,探索具有复杂化学性质的药物的目标蛋白是一项艰巨的任务(Whitebrad等人,2005年;Maryam等人,2020年;GE等人,2020年)。因此,许多研究试图通过基于大量药物和蛋白质数据的计算方法来预测DTIs(Chu等人,2019年)。这样,生物学家就可以获得可靠的候选药物-蛋白质对,以降低随后用于DTI鉴定的湿实验室实验(wet-lab experiments)的时间和金钱成本(wan等人,2019年)。
早期的DTI预测方法可以分为两类,即对接模拟方法和基于配体的方法(Cheng等人,2007年;Keiser等人,2007年)。基于对接的方法需要模拟蛋白质的三维结构,这是非常耗时的。蛋白质结构信息的缺失也限制了这些方法的应用范围。基于配体的方法需要用一组已知配体的蛋白质来查询配体。然而,这种方法可能在少量已知配体的情况下表现得不像预期的那样好。
早期的DTI预测方法可以分为两类,即对接模拟方法和基于配体的方法(Cheng等人,2007年;Keiser等人,2007年)。基于对接的方法需要模拟蛋白质的三维结构,这是非常耗时的。蛋白质结构信息的缺失也限制了这些方法的应用范围。基于配体的方法需要用一组已知配体的蛋白质来查询配体。然而,这种方法可能在少量已知配体的情况下表现得不像预期的那样好。对于蛋白质,可以通过分析从药物-蛋白质异质网络中提取的多个信息源来计算每个药物-靶向对之间的相互作用倾向(丁等人,2014年)。例如,罗等人。应用奇异值分解(SVD)(Klema和Laub,1980)从多个视角整合药物和靶标特征,并基于药物-靶标异质网络预测新的DTI(Luo等人,2017年)。这种基于网络的方法被命名为DTINet。Ezzat et al.。开发了一种基于图形正则化的矩阵分解方法GRMF,以预测潜在的DTI(Ezzat等人,2016年)。稀疏DTI矩阵通过其邻居推断可能的毒品目标来丰富。更密集的DTI矩阵有助于GRMF提高预测结果的准确性。然而,这两种基于基质分解的浅层方法未能深入探索药物与靶蛋白之间的潜在联系。Xuan等人。提出了一种基于梯度增强决策树(GBDT)的集成学习方法NGDTP(Ke等人,2017年),以探索药物的潜在靶点(Xuan等人,2020年)。他们通过整合多种类型的药物(蛋白质)相似性来构建药物(蛋白质)特征向量,但忽略了药物和靶标的自身属性,如化学指纹和氨基酸序列。Huang等人试图用名为MolTrans的变压器(Vaswani等人,2017)模型来解决这个DTI预测问题(Huang等人,2020)。与浅层方法相比,基于深度学习的方法可以更深入地保证药物与目标蛋白之间的关联。尽管如此,已知和未知的DTI之间仍然存在类别不平衡的问题。为了用平衡的数据训练MolTran,大量未知的DTI被丢弃,从而浪费了大量有价值的信息。
此外,上述方法都将DTI预测视为单标签二分类任务(已知DTI为正样本,未知DTI为负样本)。它使不同药物-靶点对的预测独立进行。然而,由于药物的化学性质和功能应该是一致的,所以化学结构相似的药物也应该有类似的结合靶点或治疗类似的疾病。如果不考虑药物与蛋白质之间的复杂配伍关系以及药物与蛋白质和疾病之间的互斥关系,单独预测可能会将相互排斥的药物视为相容的,这可能会导致后续治疗中对药物的严重误判。类似的观察也适用于蛋白质和疾病之间的关系。因此,不仅要考虑药物的标记物(药物的靶蛋白)之间的相关性,而且要保持药物与蛋白质空间之间的相关性一致性。
为此,我们试图预测潜在的DTI,同时保持药物的化学性质和功能之间的相关性一致性。我们从几个相关的公共数据库中提取了DTI预测数据集。通过构建药物-蛋白质-疾病的异质网络,将药物的化学指纹和适应症、蛋白质的氨基酸序列和已知的药物-靶点相互作用整合在一起。为了考虑不同空间药物之间的相关性,我们将DTI预测任务转化为一个多标签分类任务。具体地说,在蛋白质空间中将目标蛋白质作为药物的特征,在疾病空间中将适应症作为药物的标记物。构造了一个基于多层感知器(MLP)的编码器(Riedmiler and Lernen,2014),将药物的特征向量从原始特征空间投影到嵌入空间。利用解码器将特征从嵌入空间投影到标签空间。
我们根据三种药物的原始特征计算了三种药物的相似度、靶蛋白和适应症。通过在预测过程中最小化这些相似性之间的误差,保持了药物空间、蛋白质空间和疾病空间中药物的相关性一致性。通过这种方式,我们保持了药物自身属性和功能的一致性。我们将这种基于自动编码器的特征选择方法命名为AEFS。
2 材料和方法
本研究的主要目的是通过分析药物和蛋白质的属性以及药物、蛋白质和疾病之间的多重关联来预测潜在的DTI。分别计算了基于其化学指纹的药物相似度、基于其氨基酸序列的蛋白质相似度、基于其相关药物的疾病相似度(图1(A))。通过这些计算,构建了药物网络、蛋白质网络和疾病网络,然后通过已知的药物-靶点相互作用和已知的药物-疾病关联将其连接起来(图1(B))。一个自动编码器(Hinton and Salakhutdinov,2006)是在相关一致性的约束下建立的,以预测药物可能的目标蛋白(图1©)。
2.1 Dataset
我们使用的数据集是从几个公共数据库中提取的。DrugBank(Wishart等人,2018年)包含药物的分子结构、目标蛋白和其他信息。UniProt(Consortium,2019)是一个蛋白质相关数据库,报告了从文献和实验中获得的大量蛋白质信息。MalaCard(RapPaport等人,2017年)是一个收集人类疾病症状和相关药物的数据库。
我们从DrugBank中提取了所有化学药物的化学结构信息(简化的分子输入行输入规范,SMILES)(Anderson等,1987)和所有化学药物的靶标信息。UniProt检索蛋白质序列信息,MalaCard检索药物适应症。我们最终使用的数据集涉及1,307种药物、1,996种蛋白质、3,469种疾病,包含7,024个观察到的药物-靶相互作用和8,326个已知的药物-疾病关联。
2.2 相似度计算
2.2.1 药物相似度和疾病相似度
设表示数据集中的m种药物,m种药物的扩展连接指纹(ECFPs)(Rogers and Hahn,2010)矩阵可以基于它们的来构建。和r_, 之间的余弦相似度(Jeon等人,2019年)定义为:
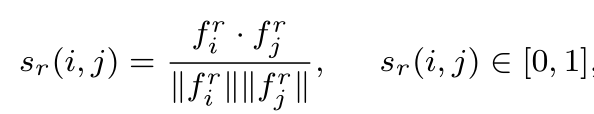
其中和分别表示r_和r_的指纹。越接近1,和越相似。由此得到药物相似度矩阵:S_r \in R^{m \times m。设表示n种疾病,根据每种疾病的相关药物,可以计算出与药物相似的疾病相似度矩阵。
2.2.2 蛋白质相似性
设表示q个蛋白质,基于其序列的蛋白质之间的相似性可以用Smith-Waterman(Crooks et al.,2005)算法和进行评分。设表示得分矩阵,q个蛋白质的相似度矩阵按公式(2)归一化计算: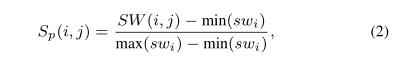
其中是和p_之间的Smith-Waterman分数,和分别表示和其他蛋白质之间的最高和最低分数。
2.3 药物-蛋白质-疾病异构网络的构建
为了整合多种类型的内部联系(即药物相似、蛋白质相似和疾病相似)和相互联系(即已知的DTIs和药物-疾病关联),我们构建了一个药物-蛋白质-疾病的异构网络。首先,我们为数据集中的药物构建了一个网络,称为。中m个药物节点之间的边由药物相似度加权。类似地,分别构建了蛋白质网络和疾病网络。
根据已知的DTIs,我们将和连接起来。具体地说,我们为每个可以相互作用的药物-蛋白质对增加了一条边。从而可以得到和之间的邻接矩阵,其中,如果和p_之间存在已知的相互作用,则,否则为0。类似地,我们还连接了和,得到了邻接矩阵,这样就可以构建一个药物-蛋白质-疾病的异质网络,如图1(b)所示。

图1.AEFS的流程图。(a)三类相似性的计算。(b)构建药物-蛋白质-疾病异质网络。©基于空间一致性约束的DTI预测自动编码器模型。
2.4 基于空间一致性约束的自动编码器
2.4.1 Autoencoder模型
为了根据药物的指纹和适应症预测潜在的DTI,构建了一个自动编码器模型,如图2所示。
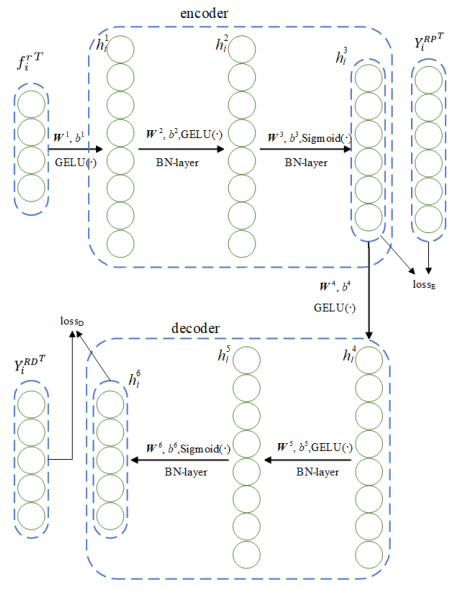
图2.基于空间一致性约束的自动编码器。的指纹是编码器的输入。编码器的输出表示和每个蛋白质之间的预测相互作用分数,并且被视为解码器的输入。解码器的输出是和每种疾病之间的预测关联分数。
具体地说,的指纹作为药物空间(原始特征空间)中的特征被用作编码器的输入。通过这两个基于MLP层的编码器,被投影到嵌入空间(蛋白质空间)。蛋白质空间中的特征向量可以计算如下:
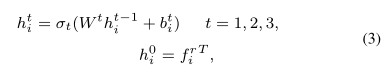
其中,、、和分别是第层的输出、激活函数、权重矩阵和偏置。和是函数(Hendrycks和Gimpel,2016年),是函数。
通过解码器将在蛋白质空间的特征向量投影到疾病空间。通过公式(4)可以得到与每种疾病之间的关联分数向量。
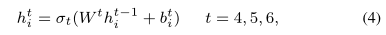
和为函数,为函数。此外,为了避免过度拟合,在每个MLP层之后添加批归一化(BN)层(Santurkar等人,2018年),以将输出向量调整为高斯分布。
2.4.2 优化器
AEFS中encoder和decoder的loss可以定义如下:
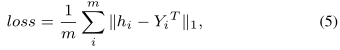
其中,编码器,,解码器,。表示通过编码器获得的与蛋白质之间的相互作用分数,存储由解码器预测的与疾病之间的关联分数。和分别包含与蛋白质和疾病之间的真实联系。通过转置操作,和被重塑为列向量。由于DTIs和药物与疾病关联的稀疏性,我们基于L1范数来度量编码器和解码器的损失。
未被观察到的靶蛋白和药物适应症导致了的特征缺失和的标签缺失。如果用这些不可靠的矩阵来优化AEFS,蛋白质空间和疾病空间中药物之间的相关性可能会改变。
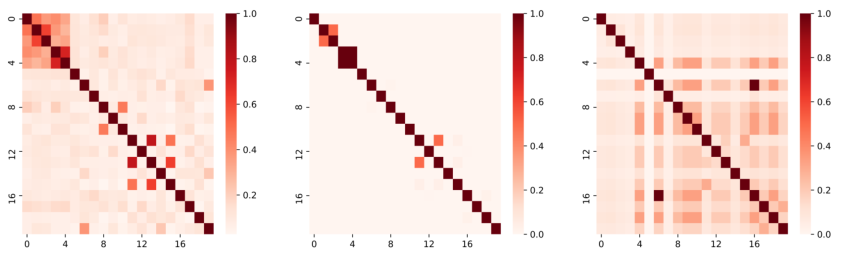
图 3. 分别在药物空间(左)、蛋白质空间(中)和疾病空间(右)中的 20 种药物之间的相关性。这 20 种药物是从数据集中随机抽取的。颜色越深,药物之间的相关性越强。蛋白质空间的相关性明显弱于药物空间和疾病空间的相关性。
如图3所示,分别根据药物化学指纹图谱、靶蛋白和药物适应证,分别计算了三个空间中20种药物(随机抽取自我们的数据集)之间的相关性(可以用药物间的相似度来衡量)。显然,这 20 种药物的相关性在这些空间中是不同的。假设药物的化学性质和功能应该是一致的,那么三个空间中的药物之间的相似性也应该是一致的。对于药物而言,其化学指纹图谱当然是确定的,而由于药物的未观察到的靶点和未知的适应症,其靶蛋白和适应症的信息很可能是不完整的。这些信息的不完备性可能会导致不同空间中药物之间的相关性发生变化。因此,保持药物的功能相关性与自身属性相关性是有意义的,也是至关重要的。为了在预测过程中保持药物之间的相关性一致性,对编码器和解码器的损失函数进行了如下扩展:
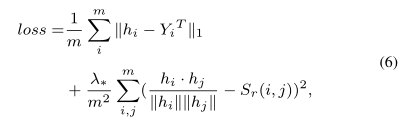
其中用于编码器,用于解码器。方程式(6)中的第二项是药物空间和蛋白质(疾病)空间之间药物相似性的损失。前者是基于编码器(解码器)的输出计算出的蛋白质(疾病)空间中和r_之间的余弦相似度。后者是两种药物在药物空间中的余弦相似度。和λ_是调整损失项贡献的参数。
此外,蛋白质和疾病的相关性也应该在药物空间中保持不变。因此,损失函数最终定义如下:
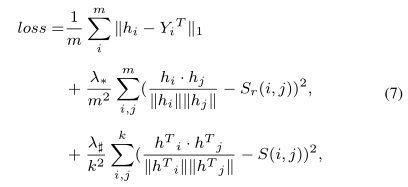
其中,,,用于编码器,,,用于解码器。以编码器为例,公式(7)中的第三项是根据编码器的输出计算的蛋白质相似度损失。与每种药物之间的相互作用分数是通过转置DTI分数矩阵得到的。类似地,可以计算药物和疾病空间之间的疾病相似度损失。药物与靶蛋白的相互作用得分矩阵可以通过最小化编码器和解码器的损失得到。
3 实验和讨论
3.1 评估指标
为了评估算法的性能,应用了 10 倍交叉验证(Luo 等人,2016 年;Wei 和 Liu,2019 年)。数据集中共有 1,307 种药物被随机分为 10 组大小相等。每组依次作为测试集,其余9组用于训练模型。在预测了所有药物蛋白质对的相互作用分数后,样品(即药物-蛋白质对)根据它们的分数按降序排序。正样本(已知 DTIs)排名越高,性能越好。此外,为了证明AEFS的鲁棒性,我们还在Luo等人(Luo等人,2017)提供的另一个数据集上进行了交叉验证。罗的数据集包含 708 种药物、1,512 种蛋白质、5,603 种疾病、1,923 种已知的 DTIs 和 199,214 种已知的药物-疾病关联。
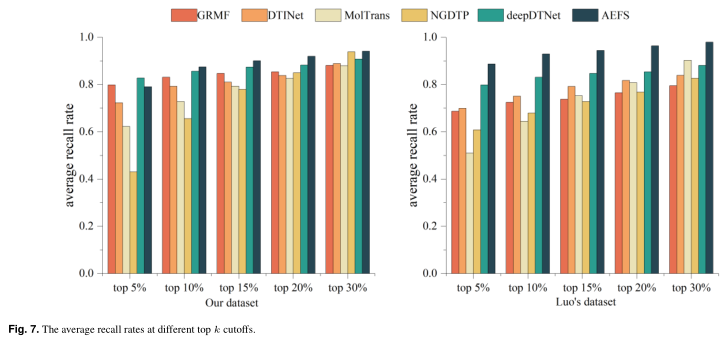
使用受试者工作特征曲线下面积(AUROC)(Haian-Tilaki,2013)来评估该预测方法的性能。此外,AUPR(精度-召回曲线下的面积)(Davis和Goadrich,2006)通常被认为是对不平衡数据更有信息量的评估指标(魏等人,2020)。因此,还构建了一条准确率-召回率曲线来评估该方法。为了从统计学角度证明AEFS的优越性能,还根据数据集中每种药物的AUROC和AUPR进行了Wilcoxon检验(Gehan,1965)。交互得分较高的候选DTI通常由生物学家通过湿法实验室实验进行鉴定。因此,收集了每种方法的前k个(5%、10%、15%、……、30%)候选样本的平均召回率,以揭示该模型对阳性样本的识别能力。此外,选择平均覆盖率作为另一个度量来说明这些方法需要多少步骤才能识别数据集中的所有已知DTIs。对于每种药物,当其召回率达到1时,覆盖率的值等于查询样本的数量。
3.2 AEFS 在考虑和不考虑相关一致性的情况下的性能
图4展示了我们的数据集在不同(选自)下测试的AEFS的性能。
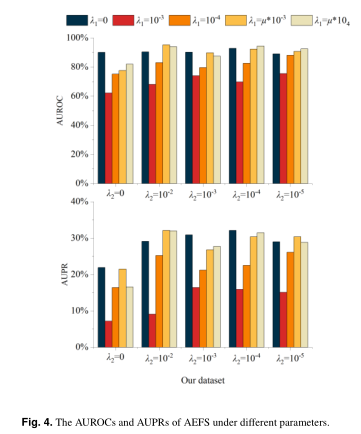
为了清楚地显示不同λ的值对结果的影响,我们设置了,。这是一个烧蚀实验,验证了当或设置为不同的值时,不同空间中的节点之间保持相关性一致性的必要性。如图4所示,通过保持药物空间和疾病空间之间的相关性一致性(即),AEFS表现得更好。请注意,当从更改时,它的性能不佳。这可能是由于药物-靶相互作用矩阵稀疏所致。为了抑制稀疏性的负面影响,我们在编码器的损失函数中增加了惩罚项。
表示矩阵的稀疏性,可通过公式(8)计算:
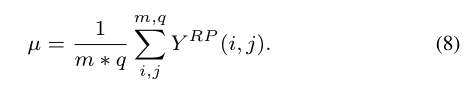
因此,编码器的损失函数可以描述如下:
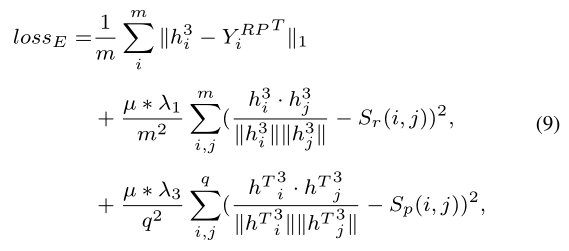
最后,当,,时,AEFS获得最佳性能。
3.3 与其他方法的比较
3.3.1 对比方法和参数设置
将AEFS与几种最先进的DTIs预测方法进行了比较,这些方法包括GRMF(Ezzat等人,2016)、DTINet(Luo等人,2017)、MolTrans(Huang等人,2020)、NGDTP(Xuan等人,2020)和deepDTNet(曾等人,2020)。
每种比较方法中的超参数都是从相关文献中推荐的范围中选取的。根据实验结果,我们在GRMF中设置了,。将DTINet中随机游走的重新开始概率设置为,,。对于NGDTP,我们在矩阵分解步骤中设置了,,,在GBDT模型中设置了num_{leaves=80,。至于MolTrans,,,,.。在deepDTNet中,我们设置,,,。
AEFS由Pytorch在GPU设备(NVIDIA GeForce GTX 1080Ti)上进行训练和优化。激活函数选自几个表现良好的函数,包括和。
编码器和解码器的深度从中选择。每个隐藏层的大小选自。批次大小选自范围。学习率选自。根据AEFS在不同参数下的AUROC和AUPR,我们最终选择Gelu和Sigmoid作为激活函数,并设置,,,.
3.3.2 实验对比
每种方法的ROC曲线和PR曲线如图5所示。
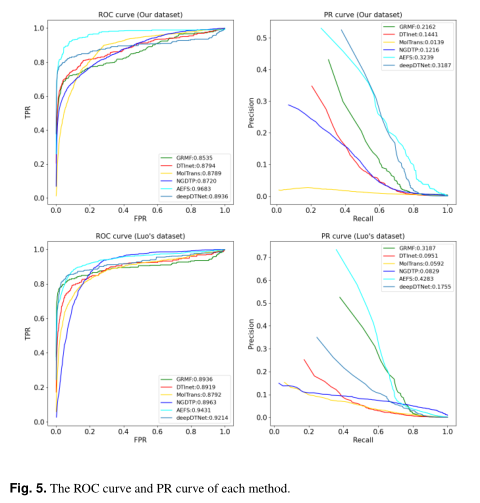
在我们的数据集上,AEFS获得了最好的性能(AUROC=96.8%,aupr=32.4%),AUROC比第二种方法DeepDTNET提高了7.47%,Aupr提高了0.52%,AUROC和Aupr分别比DTINet提高了8.9%和17.9%。MolTrans的AUROC和AUPR分别为87.9%和1.4%,AUROC和AUPR分别比AEFS低8.9%和31.0%。在我们的数据集中(正样本:负样本≈1:370)存在严重的类不平衡,它只能使用与正样本相同数量的负样本来训练模型,这可能会降低MolTrans的性能。大量可能包含有价值信息的负片样本被丢弃。尽管NGDTP(比AEFS低9.6%的AUROC和20.2%的AUPRR)是基于浅层模型的,但集成学习的思想使其能够完全利用负样本。NGDTP的AUPR比MolTrans高10.77%。与GRMF相比,AEFS的AUROC和AUPR分别为11.5%和10.8%。GRMF算法性能较差的原因可能不仅是浅层模型学习能力差,更重要的是忽略了药物节点和蛋白质节点的自身属性。
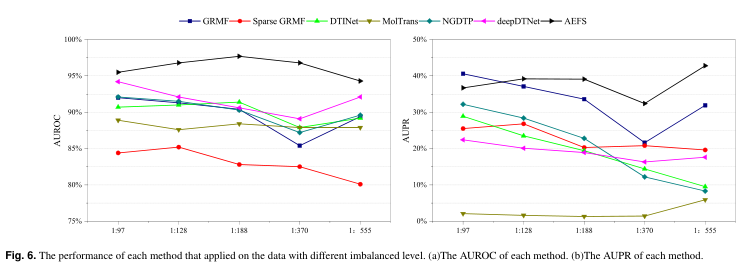
为了验证AEFS对于不同不平衡水平的不平衡数据是否具有健壮性,我们分别去除了已知相互作用药物少于1、2和3的蛋白质。通过这种方式,我们更改了数据集的不平衡级别(正样本:负样本≈分别为1:188、1:128和1:97)。罗的数据集更为稀疏,正负样本比例为1:555。GRMF通过推断更密集的DTI相互作用矩阵改变了正负样本之间的比例。因此,我们还展示了GRMF在没有推理的情况下的性能,并将其标记为稀疏GRMF。如图6所示,AEFS的综合性能最好,受类不平衡的影响较小。GRMF推导出的密集DTI矩阵使其具有相对稳健的性能。然而,当DTI矩阵过于稀疏时,GRMF很难获得足够的邻域信息来推断出高质量的DTI矩阵。
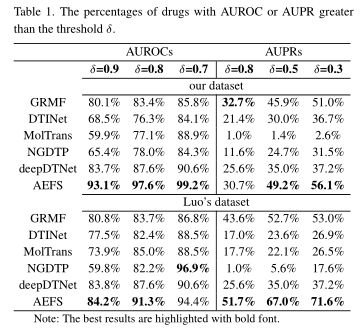
收集了每种药物上每种预测方法的性能,AUROC或AUPR大于阈值δ的药物的百分比如表1所示。为了从统计学角度验证AEFS在AUROC和AUPR上的性能,进行了Wilcoxon检验。具体地说,对于数据集中的每种药物,我们根据不同DTI预测方法下每个目标的预测分数来计算其AUROC和AUPR。根据每种方法的AUROC表和AUPR表,用Wilcoxon检验计算AEFS与各比较方法之间的p值。结果如表2所示,这表明在p值阈值为0.05的情况下,AEFS在AUROC和AUPR方面都明显优于其他方法。
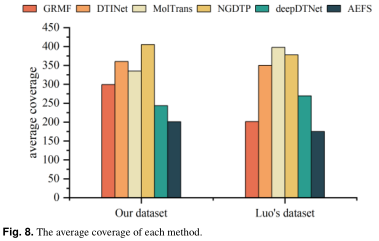
召回率揭示了该模型挖掘潜在药物蛋白相互作用的能力。Coverage表示模型查找所有阳性样本所需的步骤。使用不同的前k(5%、10%、…、30%)候选DTI,收集每种方法的平均召回率。召回率越高,识别出的DTI越多。如图7所示,AEFS在这两个数据集上取得了最好的性能。每种方法的平均覆盖率如图8所示,覆盖率越低,模型找到所有潜在DTI的速度就越快。根据图8,AEFS比所有其他方法更能有效地发现潜在的DTI。
3.4 预测新的 DTIs
为了评估AEFS的效用,我们使用数据集中所有已知的DTIs来训练AEFS,并利用它来预测所有药物的靶蛋白。AEFS预测的所有药物的前30个候选蛋白及其相互作用评分列在Supplementary ST1中。我们给出了表3中相互作用得分最高的20对药物靶点。为了验证这个预测,我们查阅了几个公共数据库寻找证据。
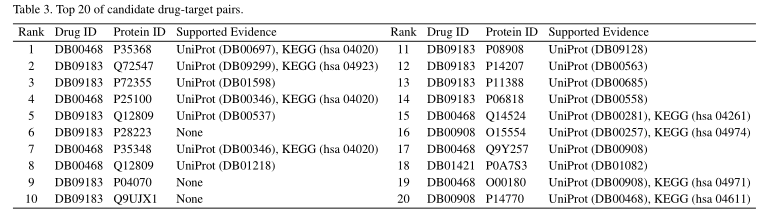
UniProt数据库报告了蛋白质的相关药物。基于相似的药物通常可以与相同的蛋白质相互作用的假设,我们试图用这个数据库为我们的预测找到支持。例如,DB00908和DB00684都被用作抗疟药物,并且DB00684和P14770之间存在已知的相互作用。因此,DB00908和P14770之间可能存在未知的相互作用。如表3所示,UniProt可以推断出20个候选dti中的17个。
KEGG途径数据库(Du等人,2014)存储蛋白质代谢途径的数据。同一途径中的蛋白质相互影响,共同表达特定的生物学功能。因此,如果一种药物能与一种蛋白质相互作用,它也可能影响与该蛋白质同一途径的其他蛋白质的表达。例如,DB00468和P35368都与钙信号通路(hsa04020)有关,因此它们之间可能存在潜在的相互作用。如表3所示,该数据库可以支持20个候选DTI中的8个。
4 结论
在这项研究中,提出了一种基于自动编码器的深度学习方法AEFS来预测潜在的DTI。构建药物-蛋白质-疾病异构网络,集成药物的化学性质、结合信息和临床信息。AEFS将药物的目标蛋白质作为其在蛋白质空间中的特征,将适应症作为其在疾病空间中的标签,利用药物的适应症信息来推断目标蛋白质。通过将DTI从传统的单标记分类任务转化为多标记分类任务,在药物-蛋白质-疾病空间中保持药物相关性的一致性,以保持药物的化学性质和功能的一致性。此外,与浅层模型相比,基于神经网络的自动编码器使得该模型在探索药物和靶蛋白之间的深层次未知关联方面更加强大。
实验结果表明,AEFS预测方法明显优于现有的几种DTI预测方法,也是一种稳健的非平衡数据DTI预测方法。经验证,AEFS的预测结果包含了许多实际的DTI。结果表明,对于生物学家来说,AEFS是一个有吸引力的选择,用于筛选可靠的候选DTI,用于随后的湿法实验室实验,以识别实际的药物-靶点相互作用。