基于综合相似性度量和双随机游走算法的药物定位
摘要:
**动机:**药物重新定位旨在为现有药物确定新的适应症,为减少传统药物开发的总时间和成本提供了一个有前途的选择。基于药物和疾病之间的相似性,已经提出了许多药物重新定位的计算策略。目前的研究通常只使用与药物相关的特性(例如,在计算药物或疾病相似性时,不考虑已知的药物-疾病关联信息对相似性度量的影响,而仅计算与疾病相关的属性(例如,表型)或仅计算与疾病相关的属性(例如,化学结构)。
结果:基于相似药物通常与相似疾病相关,反之亦然的假设,我们提出了一种新的计算方法MBiRW,该方法利用一些综合相似性度量和双随机游走(BiRW)算法来识别给定药物的潜在新适应症。通过将药物或疾病特征信息与已知的药物-疾病关联相结合,首次提出了综合相似度来计算药物和疾病的相似度。然后构建药物相似网络和疾病相似网络,并将它们合并成一个具有已知药物-疾病相互作用的异质网络。基于药物-疾病异构网络,采用BiRW算法预测新的潜在药物-疾病关联。在不同数据集上的计算实验结果表明,该方法具有可靠的预测性能,并优于目前的几种计算药物定位方法。此外,对五种选定药物的案例研究进一步证实了我们的方法在实际应用中发现潜在药物适应症的优越性能。
可用性和实施:http://github.com//bioinfomaticsCSU/MBiRW.
介绍
在过去的几十年里,基因组学、生命科学和技术取得了令人印象深刻的进步。然而,药物发现过程仍然是耗时、有风险和极其昂贵的(Li等人,2015年)。即使在研发投入不断增加的情况下,美国批准的新药数量仍在不断增加。
食品和药物管理局(FDA)自20世纪90年代末以来一直在下降(Grabowski等人,2004年)。鉴于传统药物发现面临的挑战,为现有药物确定新的适应症,也称为药物重新定位或药物再利用,吸引了制药业和研究界越来越多的兴趣(Hurle等人,2013年)。药物重新定位可以加快药物开发的进程,降低风险,因为重新定位的候选者已经通过了从头开始药物发现和开发的所有必要测试。因此,药物重新定位已成为一项重大战略,并在药物发现和开发中发挥着关键作用。此外,几种成功的重新定位药物(如西地那非、沙利度胺、雷洛昔芬)为其专利持有人或公司创造了历史上的高收入(Ashburn等人,2004年)。
药物重新定位的目标是为现有药物寻找潜在的新用途,并将新发现的药物应用于治疗药物原定疾病以外的疾病(Shim和Liu,2014)。最近,已经提出了一些计算方法来预测药物重新定位的直接药物-疾病关联。例如,Chiang和Butte(2009)开发了一种基于网络的、基于内疚的关联方法,以预测潜在的药物-疾病关联。这一方法论推荐了一种药物的新用法,其基础是假设如果两种疾病有一些相似的治疗方案,那么只对两种疾病中的一种使用的药物也可以用于另一种疾病。然而,这种方法既偏向于用途多样的旧药,也偏向于治疗多样化的疾病。Gottlieb等人。(2011)进行了多种药物-药物和疾病-疾病相似性度量来构建判别特征,并实现了一种名为预测的分类算法来预测新药适应症。吴等人。(2013)在已知疾病-基因和药物-靶点关系的基础上构建了加权的疾病和药物异质网络,并应用网络聚类来识别候选药物。Wang等人。(2014)提出了一种新的异构网络模型,该模型将药物定位和目标预测集成到一个统一的计算框架中。在该框架的基础上,开发了一种迭代算法来对每种疾病的候选药物进行排序。
玛特、�ı、内兹等人。(2015)开发了一种基于网络的优先排序方法,名为DrugNet,它同时整合有关疾病、药物和目标的信息,以执行药物-疾病和疾病-药物优先排序。Chen等人。(2015)将药物-疾病关联预测问题制定为推荐首选药物的疾病,并采用现有的两种推荐方法–PROSTS和HEATS,直接推断药物-疾病关联。
这些方法中的大多数通常通过利用药物相似性和疾病相似性来进行药物疾病预测,而相似性度量通常基于一些与药物或疾病相关的重要属性。然而,以往的研究很少利用数据集上已知的药物-疾病相互作用信息来定义相似性度量,还可以利用这些信息来改进相似性度量。
在这项研究中,我们提出了一种新的预测方法MBiRW,该方法采用有效的机制来分别度量药物和疾病的相似度,并应用双向随机游走(BiRW)算法来预测现有药物的潜在适应症。在新的相似性度量中,基于药物或疾病相关属性计算的相似性在两个方面进行了改进。首先,由于先前的研究(Van Driel等人,2006年;Vanunu等人,2010年;Wang等人,2014)发现弱相似性为交互推理提供的信息很少,我们通过相关分析调整了那些对药物疾病预测没有信息的弱相似性。其次,如果两种药物具有共同的适应症,或者同时存在其他具有共同适应症的药物,则认为这两种药物更相似。药物和疾病根据其共享的共同点进行聚类,并调整属于同一聚类的药物或疾病的相似度。
此外,由于BiRW算法在疾病基因优先排序方面比其他随机游走方法具有更好的性能,因此本研究采用BiRW算法(Xie等人,2012)来预测药物和疾病之间的潜在关联。
此外,我们还采用常用的度量标准对MBiRW在不同数据集上的性能进行了比较和评估。
实验结果表明,MBiRW具有更好的发现药物潜在疾病适应症的能力。
材料与方法
在这一部分中,首先介绍了本研究使用的数据集。
然后,基于相似药物往往指示相似疾病的假设,提出了一种将综合相似性度量与双向随机游走相结合的新方法(简称MBiRW),用于预测药物与疾病之间的潜在关联。
一般说来,MBiRW的总体预测过程包括以下三个步骤。基于收集到的数据集,首先采用综合相似性度量方法对药物和疾病进行相似性度量。在此基础上,构建了由药物相似网络、疾病相似网络和药物-疾病相互作用组成的异构网络。最后,在异构网络的基础上,实现了基于BiRW的药物候选疾病排序。
数据集
本研究中使用的金标准数据集取自Gottlieb等人的补充材料。(2011),它从多个数据来源收集了药物和疾病之间的全面联系。对于这个数据集,有1933个已知的药物-疾病关联,涉及在DrugBank注册的593种药物(Wishart等人,2008年)和在线孟德尔遗传人类(OMIM)(Hamosh等人,2002年)中列出的313种疾病。
相似性度量
相似度量方法可以简单描述如下:首先,根据一些药物相关特性和疾病相关特性,分别计算药物相似度Simrp和疾病相似度Simdp;
其次,对上述计算出的各种相似值的预测能力进行分析评价,并根据分析结果对这些相似值进行调整,得到新药相似度Simrl和疾病相似度Simdl;
最后,根据已知的药物-疾病相关性分别对药物和疾病进行聚类,然后根据聚类结果对Simrl和Simdl进行进一步改进,得到药物的Simr和疾病的Simd的综合相似度量。
药物相似度测量
根据以上描述,药物之间的相似度可以分为三个步骤进行测量。
步骤1:基于药物相关性质的相似性测量。
在本研究中,根据药物的化学结构,计算药物相似度Simrp。
微笑(简化分子输入行条目规范)是一个符号来描述化学结构,规范化的微笑(威宁,1988)的所有药物都从DrugBank下载,和化学开发工具包(斯坦贝克et al ., 2006)是用于计算相似度的两种药物Tanimoto得分(Tanimoto, 1957)的2 d化学指纹。
根据Simrp,已知的药物-疾病关联信息被纳入以下步骤中,以改善药物的相似性。
第二步:相似性分析。
根据以往研究发现弱相似性提供的预测信息较少的结果,我们分析了两种药物相似Simrp与其常见疾病存在的相关性,并将不具备预测信息的相似Simrp转化为接近于零的值。
相关分析过程描述如下:
(i)将Simrp的取值范围[0,1]分为十个相等的子范围。
对于每个子范围,计算共享常见疾病的药物对的百分比。
(ii)使用Fisher-Yates shuffling (Fisher and Yates, 1938)将Simrp的所有值随机排列成药物对,形成随机的药物相似性,其计算方法与第一步相同。
总的来说,10个随机结果的平均值被用作背景信号的控制。
(iii)根据上述结果,确定相似度阈值LSim。
也就是说,相似值小于LSim的药物对的值共享常见疾病的概率较小,因此不具备预测的信息。
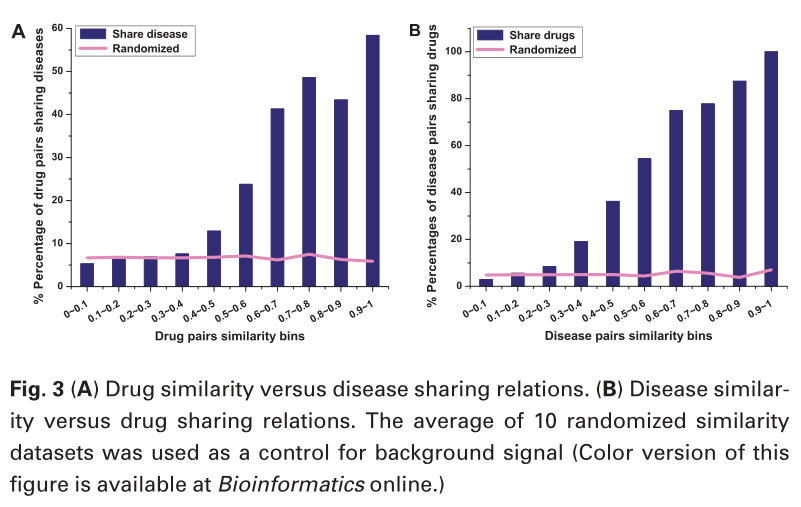
对于金标准数据集的Simrp,药物相似度相关分析结果如图3(A)所示。
在我们的工作中,我们使用Vanunu等人使用的logistic函数来调整Simrp。
(2010)修改与基因相关的疾病之间的表型相似性。
logistic函数定义如下:
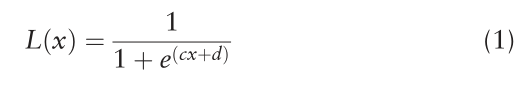
其中x为Simrp值,c和d为可以调节的参数,控制Simrp的调整。
这些小的相似值可以转化为一些接近于零的值。
同时,利用logistic函数对这些较大的相似度值进行扩展。
通过上述程序,将药物相似度mrp转化为新的相似度Simrl。
步骤3:基于已知药物-疾病相关性的药物聚类。
如前所述,药物相似度Simrl是根据相似度分析结果调整Simrp得到的。
其次,假设两种药物如果有相同的适应症就更相似,或者有其他药物同时有相同的适应症。
我们可以进一步利用已知的共享信息来提高药物相似度Simrl。
首先,我们在已知的药物-疾病关联的基础上构建了一个新的加权药物共享网络,命名为SdragNet。在SdragNet中,设SR?fr1;R2;。。。RMG表示m个药物的集合,边权重表示对应的药物对共享的常见病的数量。之后,我们对SdragNet进行聚类,以识别潜在的药物簇,并改进属于同一簇的药物之间的SIMRL。作为一种图形聚类方法,ClusterONE(尼泊尔等人,2012年)可以识别加权网络中的内聚模块,并已被用于检测有意义的药物重新定位模块(Wu等人,2013年;Yu等人,2015年)。鉴于ClusterONE在生成加权网络重叠簇方面的良好性能,本研究采用ClusterONE进行簇识别。
群集V的内聚性由ClusterONE定义如下: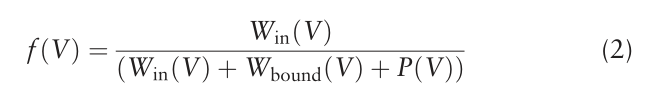
其中,WinκV?表示一组顶点V内的边的总权重,Wound§V?表示将该组连接到图的其余部分的边的总权重,P(V)是惩罚项。每个识别的集群的质量可以通过集群的内聚性来评估。
我们认为,属于同一簇的药物往往表现得更相似。这里,对于位于同一簇C中的药物ri和rj,qc表示簇C的凝聚力,将ri和rj之间的综合药物相似度Simr定义为w�Simrl,其中参数w设为α1βqc?此外,对于两种不同药物之间的综合相似度等于或大于1,我们将其替换为0.99。
疾病相似性度量
基于疾病表型的疾病相似性SIMDP是使用MimMiner(Van Driel等人,2006年)计算的,其通过计算出现在OMIM数据库中疾病的医学描述中的网格项(Lipscomb,2000)之间的相似性来测量疾病相似性。
其次,在调整药物相似性度量方法的基础上,对疾病相似性SIMDP进行了改进。首先通过考虑已知的关联信息,分析两种疾病的相似性与其常用药物的存在之间的相关性,并在相关性分析的基础上对SIMDP进行调整,得到新的疾病相似性SIMD1。然后,基于已知的药物-疾病关联,构建疾病共享网络SdiseaseNet。在SdiseaseNet中,让SD1;D2;。。。;DNG表示具有n个病种的集合,边权重表示对应病种对共享的常用药物的数量。
通过在SdiseaseNet上应用ClusterONE对疾病进行聚类,并对同一聚类内的疾病之间的相似度进行增强,以获得与药物相同的综合疾病相似度SIMD。
异构网络的建设
在上述药物和疾病相似度度量的基础上,构建了药物相似度网络和疾病相似度网络。在药物相似网络中,设R_(?)fr1;R_2;。。;RMG表示m个药物的集合。药物Ri和Rj之间的边缘由两种药物之间的综合相似度SIMR加权。在疾病相似网络中,设DüFD1;D2;。。DNG表示n种疾病的集合。疾病di和dj之间的边缘由两种疾病之间的综合相似度SIMD加权。
此外,药物与疾病的关联性可以用二部图G(V,E)来表示,其中V§G??Fr;DG;E??G?�R�D;E??G??feij,药物ri和疾病djg之间的边。如果在药物ri和疾病dj之间存在已知的关联,则将ri和dj之间的边缘的权重初始设置为1,否则,将其初始设置为0。
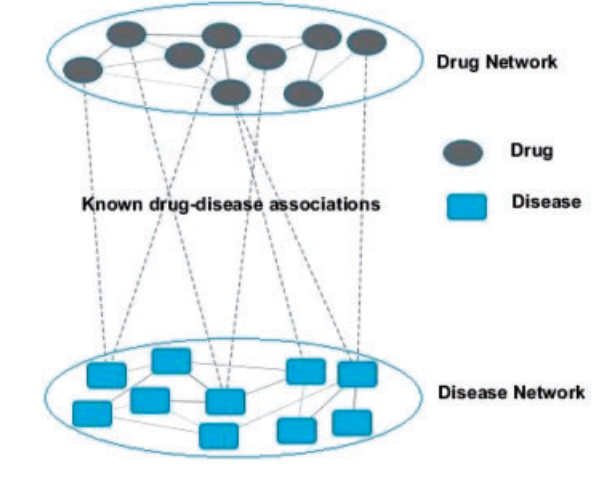
显然,药物-疾病网络可以看作是一个异构网络,它是由药物相似网络和疾病相似网络通过二分法连接而成的。药物与疾病相互作用的图表。图1显示了异构网络的原型示例。
Fig. 1 药物-疾病异质网络由药物-药物相似网络、疾病-疾病相似网络和药物-疾病相互作用组成,两个网络通过药物-疾病关联联系起来。在药物-疾病异质网络中,实线的权重表示内部相似性,虚线表示已知的药物-疾病关联
每种药物候选疾病排序的BiRW算法实现
将药物-疾病预测过程建模为同时在药物相似网络和疾病相似网络上的随机游走。考虑到不同网络的拓扑和结构特点不同,两种网络上的最佳随机游走步数可能会有所不同。因此,引入两个参数l,r作为这两个网络上左、右随机游动的最大迭代次数。迭代随机游走过程描述如下。
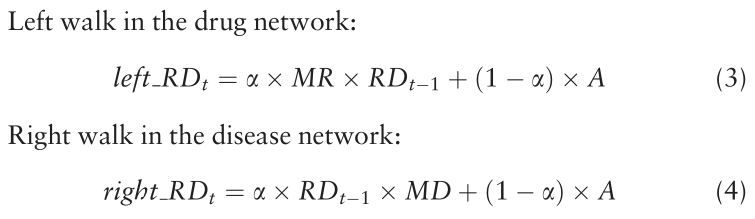
其中MRm—m、MDn—n、Am—n分别表示药物相似网络、疾病相似网络和药物-疾病关联网络的邻接矩阵,m、n分别表示药物和疾病的数量。
left_RDt和right_RDt分别表示基于药物相似网络和疾病相似网络的药物-疾病相关性预测。
元素left_RDt(i,j)和right_RDt(i,j)的值表示药物ri与疾病dj相关的概率。
在迭代过程中,RDt是每一步左步和右步的平均输出。
RDt(i, j)值越大,说明药物ri与疾病dj相关的概率越大。
用于推断潜在药物-疾病关联的完整MBiRW算法如图2所示。
实验和结果
在本节中,我们首先介绍几个用于度量各种预测方法性能的常用指标。
通过应用这些评价指标,我们在金标准数据集上对MBiRW与其他三种方法进行了综合比较。
然后,进行案例研究,以确认MBiRW的能力,寻找潜在的药物适应症。
然后,为了测试MBiRW的泛化能力,在另外两个不同的数据集上进行了系统的评价实验。


评价指标
为了系统地评价不同方法的性能,我们进行了十倍交叉验证、从头预测和独立数据集检验。
在十倍交叉验证中,金标准数据集中所有已知的药物-疾病关联被随机分为十个相同大小的亚组。
在每次交叉验证试验中,轮流取一个子集作为测试集,其余九个子集构成训练集。在进行预测后,每个关联都给出一个预测得分。
根据最终的预测得分,测试集中药物i和疾病j之间的每个已知关联相对于候选关联进行排名(所有迄今为止尚未通过实验验证与药物i相关的关联)。
指定等级阈值,TPR(真阳性)的分数是已知的正确预测的协会,玻璃钢(假阳性)的分数是未知协会预测,精度是已知关联的分数排名在等级阈值,和回忆相当于TPR。
通过改变等级阈值,我们可以计算各种TPR(真阳性率),FPR(假阳性率),精度和召回值。
然后根据这些测量值绘制受试者工作曲线(ROC)和准确召回曲线(PRC),以显示不同预测方法的性能。
考虑到预测的顶级排名结果在实践中更为重要,我们也根据顶级排名结果来衡量所有预测方法的性能,即基于不同顶级部分正确检索到的真实药物-疾病关联的数量。
通常,如果该方法能够在顶部排列更多的真关联,则被认为是更有效的。目前,许多实验性和退出性药物在药物相关数据库中没有明确的适应症,而这些药物有表明某些疾病的潜力。
在我们的研究中,我们采用从头预测试验来评估预测方法在预测新药潜在疾病(没有任何已知的相关适应症)时的有效性。
与其他方法比较
MBiRW与其他三种基于网络的预测方法进行了比较:NBI(Cheng等人,2012年)、HGBI(Wang等人,2013年)和DrugNet(Mart�ıNez等人,2015年)。NBI基于二部图上的两步扩散模型进行预测。HGBI是基于关联内疚原理和对异构图上信息流的直观解释而引入的。虽然NBI和HGBI最初是为了预测药物-靶点关联而开发的,但作者已经提到,这些预测方法也可以应用于药物-疾病网络的预测。此外,对于药物重新定位应用,它们已经在Wang等人中被用于执行药物-疾病关联预测。(2014)。DrugNet是一种基于网络的药物重新定位方法,可以进行药物-疾病和疾病-药物优先排序。本文使用相同的数据集,通过交叉验证、独立测试集和从头开始的药病预测分析,对这些方法进行了评估和比较。
药物与疾病相似性分析
在前面描述的相似度分析程序的基础上,我们首先考虑金标准数据集的已知药物-疾病关联信息,对药物相似度SIMRP进行相关性分析,结果如图3(A)所示。然后,我们用同样的方法对疾病相似性SIMDP进行相似分析,结果如图3(B)所示。
从图3中我们可以看到,相似值小于0.4的药物对表示常见疾病的概率不大,而相似值大于0.7的药物对表示常见疾病的概率很大。然后,对于药物相似度值x 2/2 0;0:4�,我们设置Lαx?�0,L(0)?0.0001和Lα0:4?<0:01,即d为log(9999),Logistic函数(1)中的c为-11。
关于图3(B),相似值低于0.3的疾病对共享药物的可能性不大,而相似值高于0.6的疾病对共享药物的可能性很大。然后,对于疾病相似度值x 2/0;0:3�,我们设置Lαx?�0,L(0)?0.0001和Lα0:3?<0:01,确定d为log(9999),c为Logistic函数(1)中的�15。最后,利用上述确定的Logistic函数进行相似度变换,分别得到调整后的药物相似度和疾病相似度。
十次交叉验证
首先通过交叉验证研究了MBiRW中使用的参数对预测性能的影响,并在补充表S1中报告了不同参数设置下的详细性能测量结果。
结果表明,参数a在0.1 ~ 0.7范围内对MBiRW的预测性能影响不大,当参数l = r时,MBiRW的预测性能更好。在本研究中,我们将参数a设为0.3,MBiRW的参数l, r设为2。
HGBI的参数(¼0.4)被设置为在(Wang et al., 2013)中使用的参数。
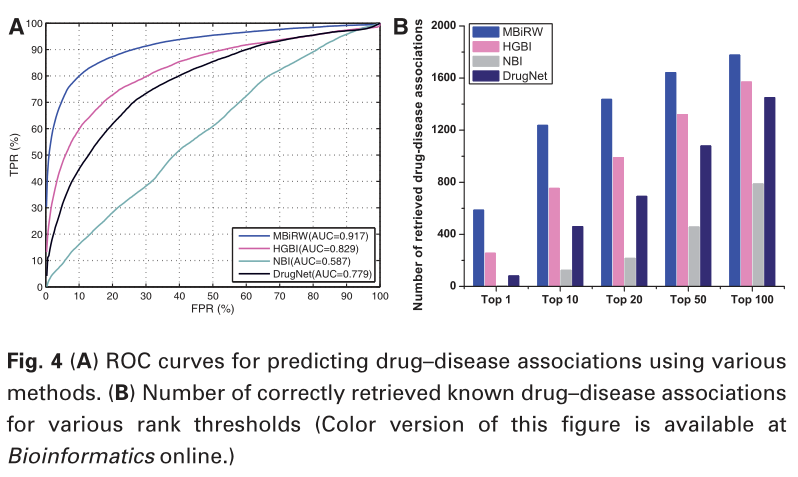
所有方法的ROC曲线评价结果和所有药物-疾病相关性的top-rank结果见图4。
需要指出的是,在每个交叉验证试验中,我们分析药物相似度和疾病相似度,并再次分析聚类药物和疾病,而不使用试验药物-疾病相关性的信息。
在AUC (ROC曲线下面积)方面,MBiRW (AUC: 0.917)优于其他方法。
此外,正确检索到的药物-疾病关联的数量如图4(B)所示。
对于指定的top-rank阈值,如果该关联的预测排名高于指定的top-rank阈值,则认为该关联是正确检索到的真实药物-疾病关联。
显然,MBiRW在聚焦于最上面的结果时,其性能明显优于其他三种方法。
例如,在1933年的真正药物-疾病关联中,有586个是基于MBiRW预测的。
排名靠前的预测在实践中尤其重要,因此MBiRW可能比其他方法更有用。
我们也通过补充图S1所示的查全率-查全率曲线报告了实验结果,MBiRW在查全率和查全率分析方面也表现出比其他方法更好的性能。
总的来说,MBiRW的预测结果表明,通过整合一些先验交互信息来调整药物和疾病的相似性可以显著提高预测的性能。
新生药物疾病预测
为了评估MBiRW预测新药潜在适应症的能力,我们进行了新药-疾病新生预测试验。
在新生预测试验中,对于每一个被询问的药物i,所有已知的药物-疾病与药物i的关联被移除。
预测方法的性能通过被移除的药物-疾病关联相对于药物候选关联的等级来评估。在金标准数据集中,每个药物至少有一个已知的相关疾病,然后我们对所有药物进行从头预测测试。
与MBiRW相比,其他三种方法也可用于预测新药的潜在疾病。
图5报道了ROC曲线和top-rank结果的实验结果。
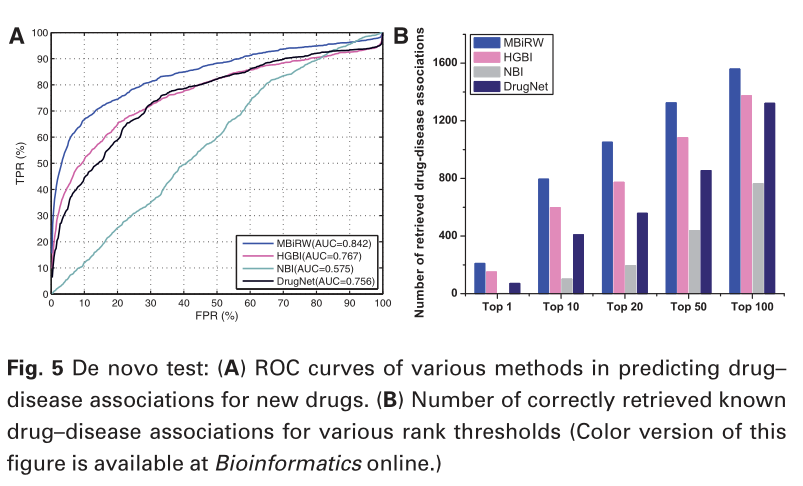
从图5可以看出,MBiRW的AUC值为0.842,在相同的实验场景下,也优于其他方法。
同时,在关注排名最高的预测结果时,在1933个已知的药物-疾病关联中,MBiRW预测了209个排名最高的。
从补充图S2的PR曲线分析结果可以看出,MBiRW的预测也是最优的。
总的来说,所有的新生预测结果表明,我们的方法可以达到优越的性能。
独立的测试集
我们还调查在独立测试集上药物-疾病关联预测的各种方法的性能。为了创建独立测试集,发表的药物-疾病关联首先来自KEGG (Kanehisa et al., 2014)数据库和文献(Mart�ınez et al., 2015)。
然后,在去除金标准数据集中存在的关联后,可以获得一个包含115种药物的144个新的药物-疾病对的独立测试集。
对独立数据集进行检验,通过在独立数据集中预测新的药物-疾病关联,来衡量所有预测方法的性能。
ROC曲线、top-rank分析和PR曲线的检测结果见补充图S3。
从这些结果中我们可以清楚地看到,MBiRW的AUC值最好,为0.831,有15个新关联排在首位。
此外,MBiRW在PR曲线方面也优于其他方法,精度最高。
所有这些结果表明MBiRW在独立测试集上取得了最好的性能,这与对金标准数据集的评价是一致的。
衡量综合相似度量的效果
综合相似测度的计算包括2.2节所述的三个步骤。
我们通过交叉验证实验比较了BiRW1、BiRW12、BiRW13和MBiRW四种方法。
在这些方法中,在相似度量中考虑了各种相似计算步骤。
对于BiRW1,第一步涉及相似性度量,这意味着只使用药物或疾病相关的属性信息来计算相似性。
对于BiRW12来说,步骤一和步骤二都是在相似度量中实现的,即步骤一所度量的相似度通过相应的相似度分析进行调整。
对于BiRW13来说,第一步和第三步都是在相似度测量中进行的,这意味着第一步测量的相似度是通过考虑已知的药物-疾病相关性来调整的。
对于MBiRW来说,这三个步骤都涉及到相似性度量。
这些方法的ROC曲线比较结果和排名靠前的结果如补充图S4所示。结果表明,BiRW1比所有其他方法都要差。BiRW1的AUC值为0.87,仅能正确检索到最上面的166个关联。而BiRW12和BiRW13的AUC值分别为0.896和0.897,这意味着通过将已知的关联信息结合到相似性度量中可以提高预测性能。MBiRW在AUC值、排名靠前的结果和PR曲线上都优于所有其他方法,进一步证明了综合相似性度量的有效性。
个案研究
在确认了我们基于交叉验证的方法的优异性能之后,这里进一步检验了MBiRW在预测新的药物-疾病关联方面的能力。为了预测药物的新适应症,金标准中所有已知的药物-疾病相互作用被用作训练集,其余在我们的金标准数据集中未知关联的药物-疾病对形成候选药物-疾病关联的集合。通过应用MBiRW方法,我们可以得到所有候选药物-疾病对的预测得分。MBiRW可以同时预测所有药物的潜在关联性。
对于每种药物,根据预测分数对候选疾病进行排名,并收集作为预测结果的前10名预测疾病。
补充表S2中列出了对所有药物的预测。
在这里,我们随机选择几种药物,并提出他们的预测进行案例研究。基于一些公共数据库、当前的临床试验和文献,预测结果得到了证实。公共数据库包括KEGG(Kanehisa等人,2014年)、DrugBank和CTD(Davis等人,2015年)。在这些数据库中,一些新增加的药物-疾病关联为我们的验证提供了基础。对于每一种选定的药物,与指定药物相关的前5种潜在疾病和证据列在补充表S3中。我们发现,一些排名靠前的预测已经得到现有研究的证实。例如,左旋多巴被预测用于治疗阿尔茨海默病,这已经在临床试验中得到了测试(临床试验.gov,2006a)。
阿霉素,用于治疗非小细胞肺癌(NSCLC),被预测用于治疗小细胞肺癌(SCLC)。这一预测得到了CTD的证实。此外,对于阿霉素,治疗前列腺癌的预测已经在临床试验(临床试验)中得到验证。
政府,2006b)。卡麦角林,被认为是高催乳素血症,被预测用于治疗移植物,这一预测已经在文献中进行了研究(Erkulwater和Pillai,1989)。金刚烷胺,用于治疗帕金森氏症,被预测用于治疗阿尔茨海默病,这种疗法已经在文献中进行了研究(Cavestro等人,2006年)。
此外,金刚烷胺衍生物美金刚是一种用于治疗阿尔茨海默病的N-甲基-D-天冬氨酸(NMDA)受体拮抗剂。这些成功的预测实例进一步证实了MBiRW具有预测药物的新疾病适应症的潜力。
在其他数据集上的实验
现有的大多数研究都是基于一个特定的数据集进行性能评估实验,而忽略了算法对不同数据集的适应性。为了使预测结果更具说服力,在另外两个数据集上进一步进行了实验。
首先,从论文(MART�ıNEZ等人,2015年)获得一个新的数据集,其包含由疾病本体(DO)术语标注的4516种疾病,在0 10 20 30 40 40 50 70 90 90 100 0 10 20 30 40 50 60 70 80 90 100 0 10 20 30 40 50 60 70 80 90 100FPR(%)TPR(%)MBiRW(Auc=0.842)HGBI(Auc=0.767)NBI(Auc=0.575)DrugNet(Auc=0.575)中获得一个新的数据集。(B)正确检索到各种等级阈值的已知药物-疾病关联的数量(此图的彩色版本可在生物信息学在线上获得。)。DrugBank和1008个已知的药物-疾病协会,这些协会都是从DrugBank派生的。
此外,本文还将DNA数据集与本文使用的金标数据集相结合,生成了另一个数据集CDataset。CdataSet包括在DrugBank注册的663种药物,OMIM数据库中列出的409种疾病,以及2352个已知的药物-疾病协会。
在DNA数据集中,药物相似性SIMRP基于解剖治疗化学(ATC)编码来度量,疾病相似性Si MDP基于疾病本体(DO)术语来度量。在CDataSet中,药物相似性SIMRP是基于化学结构来测量的,疾病相似性SIMDP是使用MimMiner基于表型来测量的。分别从十倍交叉验证和从头预测两个方面对MBiRW在DN数据集和CDataSet上的性能进行了评估。
对DNA数据集的验证
首先,我们在DNA数据集上进行了十次交叉验证来验证我们的方法的性能,实验结果如补充图S5所示。MBiRW中使用的参数与用于评估上述金标准数据集的实验中使用的参数相同。在这里,对于DNA数据集,参数设置可能不是最佳的,可以进行调整以获得最佳结果。
交叉验证结果表明,MBiRW和DrugNet的性能优于其他方法,MBiRW和DrugNet的AUC值分别为0.958和0.948。
虽然MBiRW在ROC曲线和AUC值方面仅略好于DrugNet,但在一流的分析和精度结果方面,MBiRW具有明显的优势。
此外,还在DNA数据集上进行了从头预测检验。在DNA数据集中,有550种药物至少有一个已知的关联,所以我们对这550种药物进行从头预测。预测结果载于补充图S6。与以上十倍交叉验证结果相比,所有方法在从头预测结果中的AUC值均略有提高。原因可能是因为DNA数据集包括许多只有一个已知关联的药物。在十重交叉验证中,将所有已知关联划分为10个部分,每个部分依次作为测试集,其余9个部分作为训练集。因此,存在不止一种药物,它只有一种相关疾病,被认为是没有已知关联的药物。然而,在每个从头开始的药物-疾病预测中,只删除与查询的药物有关的已知药物-疾病关联。也就是说,在从头预测中,没有已知疾病适应症的药物数量可能少于交叉验证中的10倍。
CdataSet上的验证
对于CDataSet,实现了药物-疾病关联预测的十次交叉验证实验,评估结果如补充图S7所示。结果表明,该方法获得的AUC值为0.934,且精度较高,明显优于其他方法。从排名靠前的结果可以看出,在2352个已知的药物-疾病关联中,MBiRW可以预测出887个关联位于前一个。相比之下,HGBI、NBI和DrugNet分别预测了427个、148个和84个已知关联。
每种药物在CDataset中至少有一个已知适应症,所以我们对所有药物进行从头预测,结果如补充图S8所示。与十倍交叉验证相比,我们发现在从头检验中,AUC值和排名靠前的结果都变得更差了。例如,十次交叉验证获得的AUC值达到了0.934,而在从头预测中AUC值只达到了0.842。排名前一的预测关联数为241个,显著少于十倍交叉验证的关联数。
在DNA数据集和CDataSet上的卓越性能评估结果进一步表明,MBiRW在预测潜在的药物-疾病关联方面是可靠的。通过以上实验,我们得出结论:在相似性度量中集成更多有用信息,并采用BiRW算法,可以提高预测性能。
Conclusion
药物重新定位是药物开发的一种很有前途的选择。在这项工作中,我们提出了一种新的方法,名为MBiRW,揭示了药物和疾病之间的潜在联系。本研究的主要贡献在于,通过有效整合已知的药物-疾病关联信息,设计了新的药物和疾病相似度度量方法,并基于BiRW算法进行了药物定位。相似性度量的步骤包括:根据先验的生物特征计算药物和疾病的相似度;对相似度值进行相关性分析,并根据分析结果通过Logistic函数调整相似度;基于已知的药物-疾病关联度对药物和疾病进行聚类,并基于识别出的聚类提高药物和疾病的相似度。
在计算综合相似度的基础上,构建了药物网络和疾病网络。此外,药物网络通过已知的药物-疾病协会与疾病网络相连,构成一个药物-疾病异质网络。最后,采用BiRW算法对每种药物的候选疾病进行排序,并在收集到的数据集上验证了算法的有效性。在交叉验证中,所有的实验结果都表明,与其他方法相比,我们的方法可以有效地提高预测性能。对几种药物的案例研究表明,MBiRW预测的潜在药物-疾病关联可能对生物医学研究更有用。
MBiRW是一种强大的计算方法,可以同时预测不同药物的候选疾病,也可以在没有任何已知相关疾病信息的情况下有效地预测药物的新疾病适应症。我们从交叉验证和案例研究两个方面验证了它的预测能力。虽然我们的方法的结果是有希望的,但它的局限性应该得到承认。首先,MBiRW只使用已知的药物-疾病关联信息来提高药物对和疾病对的相似度。应该指出的是,存在更多的先验生物相关信息,可以合理地利用这些信息来改进相似性度量。其次,考虑到存在大量与药物和疾病相关的目标数据,我们计划在考虑目标信息的基础上构建全面的异构药物定位网络。
References
Ashburn,T.T. et al. (2004) Drug repositioning: identifying and developing
new uses for existing drugs. Nat. Rev. Drug Discov., 3, 673–683.
Cavestro,C. et al. (2006) High prolactin levels as a worsening factor for mi-
graine. J. Headache Pain, 7, 83–89.
Chen,X. et al. (2012) Drug–target interaction prediction by random walk on
the heterogeneous network. Mol. BioSyst., 8, 1970–1978.
Chen,H. et al. (2015) Network-based inference methods for drug reposition-
ing. Comput. Math. Methods Med., 2015, 130620.
Cheng,F. et al. (2012) Prediction of drug–target interactions and drug
repositioning via network-based
inference.
PLoS Comput. Biol., 8,
e1002503.
Chiang,A.P. and Butte,A.J. (2009) Systematic evaluation of drug–disease rela-
tionships to identify leads for novel drug uses. Clin. Pharmacol. Therap.,
86, 507–510.
ClinicalTrials.gov
(2006a)
Docetaxel,
Doxorubicin, and Prednisone in
Treating Patients With Advanced Prostate Cancer That Has Not Responded
to Hormone Therapy.
ClinicalTrials.gov (2006b) Dopaminergic enhancement of learning and mem-
ory in healthy adults and patients with dementia/mild cognitive impairment.
Davis,A.P. et al. (2015) The Comparative Toxicogenomics Database’s 10th
year anniversary: update 2015. Nucleic Acids Res., 43, D914–D920.
Erkulwater,S. and Pillai,R. (1989) Amantadine and the end-stage dementia of
Alzheimer�s type. Southern Med. J., 82, 550–554.
Fisher,R.A. and Yates,F. (1938) Statistical tables for biological, agricultural
and medical research. Edinburgh: Oliver and Boyd.
Hamosh,A. et al. (2002) Online Mendelian Inheritance in Man (OMIM), a
knowledgebase of human genes and genetic disorders. Nucleic Acids Res.,
30, 52–55.
Hurle,M.R. et al. (2013) Computational drug repositioning: from data to
therapeutics. Clin. Pharmacol. Therap., 93, 335–341.
Gottlieb,A. et al. (2011) PREDICT: a method for inferring novel drug indica-
tions with application to personalized medicine. Mol. Syst. Biol., 77, 496.
Grabowski,H. (2004) Are the economics of pharmaceutical research and de-
velopment changing? Pharmacoeconomics, 22, 15–24.
Kanehisa,M. et al. (2014) Data, information, knowledge and principle: back
to metabolism in KEGG. Nucleic Acids Res., 42, D199–D205.
Li,J. et al. (2015) A survey of current trends in computational drug reposition-
ing. Brief. Bioinf., 1, 11.
Lipscomb,C.E. (2000) Medical subject headings (MeSH). Bull. Med. Libr.
Assoc., 88, 265–266.
Mart�ınez,V. et al. (2015) DrugNet: Network-based drug–disease prioritiza-
tion by integrating heterogeneous data. Artif. Intell. Med., 63, 41–49.
Nepusz,T. et al. (2012) Detecting overlapping protein complexes in protein–
protein interaction networks. Nat. Methods, 9, 471–472.
Shim,J.S. and Liu,J.O. (2014) Recent advances in drug repositioning for the
discovery of new anticancer drugs. Int. J. Biol. Sci., 10, 654.
Steinbeck,C. et al. (2006) Recent developments of the chemistry development
kit(CDK)-an open-source java library for chemo- and bioinformatics. Curr.
Pharm. Des., 12, 2111–2120.
Tanimoto,T. (1957) An Elementary Mathematical theory of Classification and
Prediction. Internal IBM Technical Report.
Wang,W. et al. (2013) Drug target predictions based on heterogeneous graph
inference. Pac. Symp. Biocomput, 18, 53–64.
Wang,W. et al. (2014) Drug repositioning by integrating target information
through a heterogeneous network model. Bioinformatics, 30, 2923–2930.
Weininger,D. (1988) SMILES, a chemical language and information system.
- Introduction to methodology and encoding rules. J. Chem. Inf. Model.,
28, 31–36.
Wishart,D.S. et al. (2008) DrugBank: a knowledgebase for drugs, drug actions
and drug targets. Nucleic Acids Res., 36, D901–D906.
Wu,C. et al. (2013) Computational drug repositioning through heterogeneous
network clustering. BMC Syst. Biol., 7, S6.
Van Driel,M.A. et al. (2006) A text-mining analysis of the human phenome.
Eur. J. Human Genet., 14, 535–542.
Vanunu,O. et al. (2010) Associating genes and protein complexes with disease
via network propagation. PLoS Comput. Biol., 6, e1000641.
Xie,M. et al. (2012) Prioritizing disease genes by bi-random walk. Adv.
Knowl. Discov. Data Mining, 2, LNCS, 7302, 292–303.
Yu,L. et al. (2015) Inferring drug–disease associations based on known protein
complexes. BMC Med. Genomics, 8, S2.